


The Importance of End-To-End Thinking During Drug Product Development - Outsourced Pharma
Throughout a drug product development program it is important to build robustness and flexibility into the manufacturing process. In this article, Dr. Kashif Ghaffar, process validation pharmacist at Pfizer's Freiburg manufacturing site, discusses what needs to be considered during product development in preparation for commercial manufacture.
End-to-end view
During early drug development there is a tendency to adopt a ‘fit-for-purpose’ strategy when producing formulations for clinical trials. This approach is understandable considering that only one in ten compounds that enter clinical trials will become registered products. Nevertheless, it is important to build in robustness for manufacture early in development and to prepare for scale-up.
There may be reasons for changing formulation or process strategy during early product development (e.g. direct compression to granulation approach). As a result, being agile through having access to a broad range of processing equipment with scale-up capability is extremely advantageous.
The use of continuous processing strategies, such as roller compaction and continuous mixing, offer great potential for batch size flexibility. For larger batch sizes you simply run the process for longer. The FDA’s ‘scale-up and post approval changes’ (SUPAC) guidelines1,2 provide a useful framework for manufacturing scale-up and it is highly beneficial if equipment of the same SUPAC class is available for development, scale-up and commercial manufacturing.

Figure 1. Ensure processing equipment is available for scale-up to larger scale manufacture
Regulatory expectations
The principles for ‘Pharmaceutical Development’, with regards to regulatory expectations, are outlined in ICH Q83 and an important element is Quality-by-Design (QbD). The basic principle behind QbD is that “quality should be built into a product with an understanding of the product and process by which it is developed and manufactured along with a knowledge of the risks involved in manufacturing the product and how best to mitigate those risks”.
QbD has been embraced by the regulatory agencies as a ‘best practice’ for pharmaceutical product development, and the FDA and EMA have recently published the conclusions of their first parallel assessment of QbD elements of marketing-authorisation applications4. An important aspect of QbD is understanding and controlling the manufacturing process for the product and the impact each process step is having on each other and at the critical quality attributes. Defining the quality-critical process parameters (e.g. air flow/temperature, rotor speed, spray rate) and then monitoring these parameters during manufacture, is a standard practice in pharmaceutical manufacture.
This mapping of the relationship between process inputs and quality critical attributes can be described as defining the ‘design space’ for a process and is an important contributing element to QbD. Gained product and process knowledge will be advantageous throughout the entire life cycle, such as in following validation activities due to equipment or raw material changes. An excellent review of QbD relating to pharmaceutical product development is provided by Yu, et al.5
A schematic representation of the QbD concept is shown in Figure 2 and the relationship of ‘Design Space’ and QbD is shown in Figure 3.
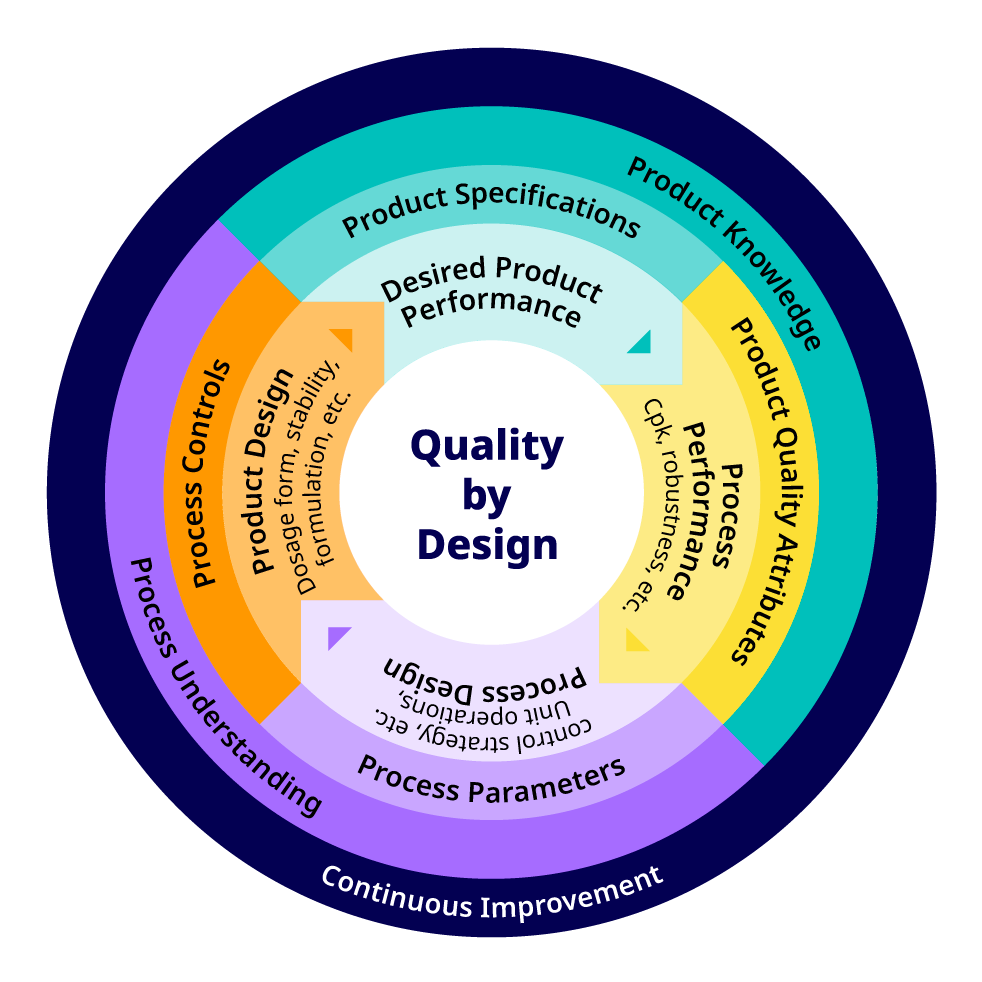
Figure 2. Quality by design (QbD) concept as presented by U.S. Food and Drug Administration (FDA) 6
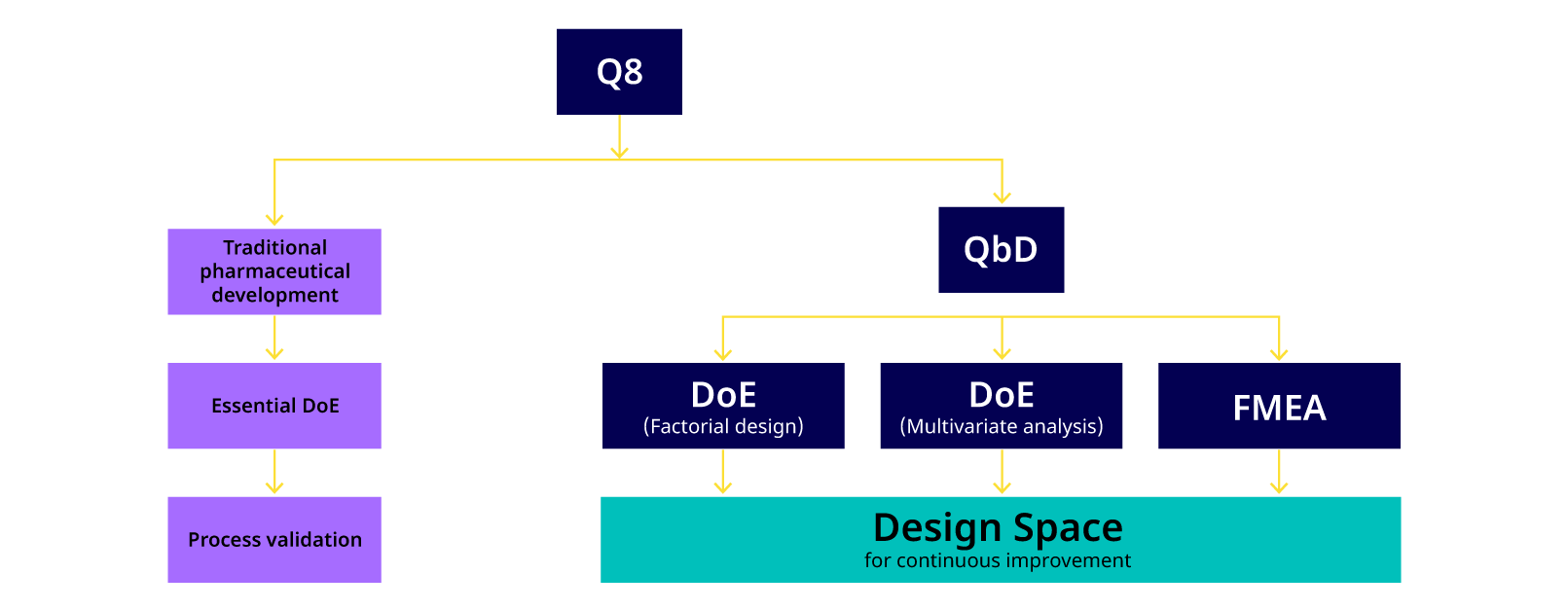
Figure 3. Current pharmaceutical development practice (Q8) versus the traditional approach
There is a close relationship between ICH Q8 (Pharmaceutical Development), Q9 (Quality Risk Management) 7 and Q10 (Pharmaceutical Quality System) 8 guidelines. These guidelines provide a structured approach from early product development to commercial product and underline the importance of knowledge management during the product life cycle. The relationship between these guidelines is shown in Figure 4.
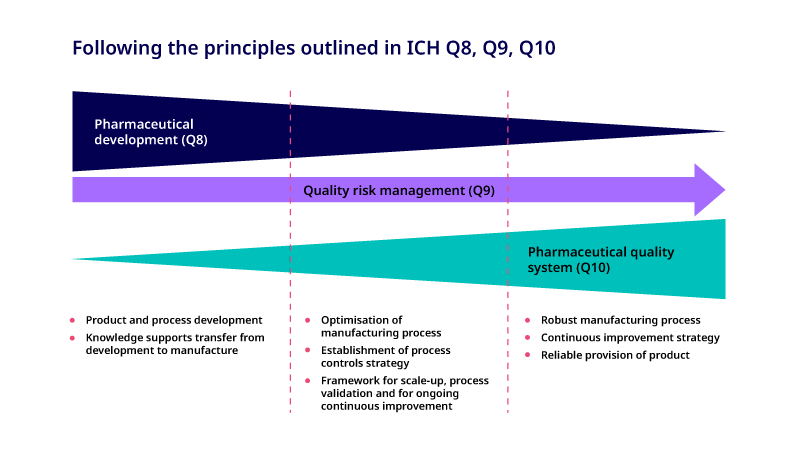
Figure 4. Schematic illustrating the relationship between ICH Q8, Q9 and Q10 throughout the drug development process
Anticipating and managing manufacturing issues
Having thorough knowledge and experience in oral sold dose manufacturing processes helps identify potential problems and facilitates the application of appropriate practices to prevent issues. For instance, drug content uniformity may be an issue during powder blending due to segregation effects, and it is important for a CDMO to have experience in managing such situations.
Other issues may include tablet ‘sticking’ to tablet punches (which can be addressed by use of non-stick coatings on tablet punches) and ‘twinning’ during tablet coating (prevented by careful control of coating parameters).
Conclusions
An end-to-end view of the product life cycle throughout product development is key. While a degree of pragmatism is expected during early stage development, it is critical to be aware of the practicalities to meet future demands for successful scale-up in due course.
Building a firm foundation early in the product life cycle is central to successful product development and for providing a robust, scalable manufacturing process for a new product.
References
-
SUPAC-IR Guidelines, 1995 (US Food and Drug Administration)
-
SUPAC-MR Guidelines, 1997 (US Food and Drug Administration)
-
ICH Guideline, Pharmaceutical Development, Q8(R2), August 2009.
-
EMA-FDA pilot program for parallel assessment of Quality-by-Design applications: lessons learnt and Q&A resulting from the first parallel assessment. EMA/430501/2013.
-
Yu, L.X., et al., Understanding Pharmaceutical Quality by Design. The AAPS Journal (2014), 16(4): 771-783.
-
https://www.qualitydigest.com/inside/twitter-ed/taking-holistic-approach-quality-design.html
-
ICH Guideline, Quality Risk Management, Q9, November 2005.
-
ICH Guideline, Pharmaceutical Quality System, Q10, June 2008.